


CD47: Eating Up Cancer and Biobucks
8,500 words

This article was initially a head-to-head comparison of Trillium vs ALX, and has been revised to focus on the greater CD47 landscape following $TRIL’s buyout. We also deep dive on $ALXO $IMAB $STTK $OSE $TRIL below.
1. What is CD47-SIRPα?

Today, most cancer immunotherapy drugs focus on the adaptive immune system. But CD47-SIRPα is exciting because of its potential as an innate immune system cancer drug, which can be used in combination with established adaptive immunotherapy drugs and chemotherapy for hard to treat cancers, with high safety & efficacy. In fact, CD47 could become an innate immunotherapy backbone, much like PD-(L)1 for the adaptive system. Macrophages, which CD47 drugs act on, are relatively understudied versus T-cells, which have received more love from both scientists and investors.
In fifty words / TL;DR
Macrophages are the innate immune system’s garbagemen. Cancer cells evade them by sounding a “don’t eat me!” signal when their CD47 ligands slot into a macrophage’s SIRPα receptor. This handshake prevents phagocytosis. When this handshake is blocked by a CD47-SIRPα drug and combined with other drugs or an active ‘eat me’ signal, macrophages can eat cancer cells.
In three-hundred words
Evasion of the immune system is a hallmark of cancer. The development of therapeutics that target these mechanisms, which cancer uses to evade the immune system, have revolutionized treatment. These drugs, referred to as immune-checkpoint inhibitors (ICIs), function by targeting inhibitory immunoreceptors. For example, antibodies targeting T-cell inhibitory receptors, such as CTLA-4 and PD-1, are able to activate the host’s adaptive immune system to attack cancer cells. A major success of ICIs has been the long term remission after discontinuation of treatment experienced by a subset of patients. This kind of long-term remission was unfathomable before the development of ICIs.
While revolutionary, existing ICIs have numerous limitations. Though immune checkpoint blockade often leads to more durable responses than other therapies, it has a low response rate, often between 10% and 30%, for most cancers. Additionally, combining existing ICIs can lead to severe treatment related adverse events (TRAE). When anti-CTLA & anti-PD1 are used in combo, up to 60% of patients experience grade 3 or higher TRAE.
Moreover, there is a lack of ICIs that target the innate immune system, with the majority of existing ICIs targeting the adaptive immune system. However, the coordinated response of the adaptive and innate immune system is required for a robust anti-tumor immune response - one that engages the best of both sides of our two immune systems - which means finding potential ‘backbone’ ICIs that target the innate immune system.
The CD47-SIRPα pathway is a critical checkpoint of the innate immune system. CD47 is commonly referred to as a “don’t-eat-me” signal since SIRPα and CD47 binding results in a signal that downregulates cell phagocytosis. CD47 is overexpressed in numerous hematological malignancies and solid tumors. This overexpression of CD47 allows cancer cells to avoid phagocytosis. The CD47-SIRPα checkpoint has emerged as the most promising innate immune checkpoint with numerous therapeutics targeting this axis in the clinic.
2. The Comprehensive CD47 Landscape

Late-September Update: Korean company PharmAbcine (KOSDAQ: 208340) is developing PMC-122, a preclinical PD-L1 + CD47 BsAb.
Press the image above to enlarge; email us for the PPT file
What to know:
We are still in the early-mid stages of a global CD47 race, complete with 36 players, 50 drug candidates, and 80+ global trials.
There is no commercialized CD47-SIRPα drug yet.
There have been two landmark CD47 acquisitions. Gilead’s first CD47 acquisition of Forty Seven ($FTSV) in 2019 for $4.9B, the first-in-class player cofounded by Stanford Professor Irv Weissman, whose lab discovered the relevance of CD47 as an target for cancer immunotherapy, put CD47 on the biotech radar. A few weeks ago, Pfizer’s acquisition of Trillium for $2.3B was announced.
There can and will be multiple winners. While Gilead/$FTSV is first-in-class with magrolimab likely to be the first commercialized, there remains ample opportunities for different players to play and win across hemes & solid cancers
Best-in-class is still too early to call. A best-in-class winner will emerge in 2022 in both heme and solids. This race is closely led by ALX Oncology (Phase 2) for solids and Trillium (Phase 2) seeking to claim Gilead’s (Phase 3) throne for hemes, followed by I-Mab, Shattuck, and a bunch more private and international players.
Most players are CD47 mAbs (monoclonal antibodies) or bsAbs (bispecific monoclonal antibodies), but fusion proteins appear to be the most promising class
SIRPα mAbs are emerging as a novel therapeutic approach, but there is still too little data to make any definitive conclusions
There are many promising private players. Leaders of the pack include ARCH, ImunneOnco, and KAHR, and many Chinese biotechs
3. Science Review
Note: This part is for more advanced readers. Skip below for due diligence.
CD47
CD47 is a ~50kDa transmembrane protein that is heavily glycosylated and expressed ubiquitously on the surface of cells. CD47 is known to bind integrin, thrombospondin-1, and SIRPα. CD47 plays a role in numerous cellular processes, including migration, adhesion, proliferation, and apoptosis. CD47 is an antiphagocytic ligand that functions a marker of self.
Cancer cells exploit this “don’t eat me” signal to evade the immune system. CD47 is overexpressed a variety of hematological cancers and solid tumors. This overexpression is clinically correlated with a poor prognosis. It is clear that CD47 plays a key role maintaining an immunosuppressive tumor microenvironment.
SIRPα
SIRPα (CD172a) is an immunoreceptor tyrosine-based inhibitory motif (ITIM) containing receptor, is an ~65 kDa inhibitory receptor that belongs to a family of cell surface receptors that is known to modulate immune responses. SIRPα is closely related to SIRPβ1 (CD172b), an activating receptor, and SIRPγ (CD172g), a nonsignaling receptor. SIRPβ2 and SIRPδ are known, but are distantly related to SIRPα and poorly characterized. SIRPα has a narrow expression profile and is found primarily on the membrane of myeloid cells, such as macrophages and myeloid dendritic cells. SIRPα helps regulate cell migration and phagocytosis.
CD47-SIRPα Signaling
Ligation of CD47 by SIRPα results in the phosphorylation of the tyrosine residue in ITIM of SIRPα. Phosphorylation of the ITIM furnishes a docking site for a tyrosine phosphatase, either SHP-1 or SHP-2 (in macrophages SHP-1 is the predominant enzyme). This recruitment of SHP-1 or SHP-2 by SIRPα, results in the transmission of inhibitory signals that downregulate phagocytosis.
General Strategy for Targeting CD47-SIRPα
The goal is to block the binding of CD47 on cancer cells to SIRPα on immune cells. By doing so, it should start to take the brakes off of the immune system, by stopping the “don’t eat me” signal that cancer cells are giving to the macrophages. While cancer cells usually have some existing prophagocytic signal, blocking CD47 & SIRPα binding is not enough to trigger phagocytosis of the cancer cells. Instead, the prophagocytic signal needs to be reinforced by labeling cancer cells with an additional “eat me” signal.
Modalities
1) Anti-CD47 mAbs:
Anti-CD47 mAbs were the first strategy used to successfully blockade CD47-SIRPα. Both fully human and humanized anti-CD47 mAbs have been developed and advanced to the clinic. Most of these agents utilize an IgG4 Fc region for multiple reasons. Most importantly, if the goal when designing the agent is to be active as a monotherapy, the Fc region needs to be active since a prophagocytic signal is required to trigger phagocytosis. However, since CD47 is ubiquitous, choosing an Fc isotype with higher affinity for certain Fc receptors will result in greater Fc receptor mediated effector function that could lead to greater off target cell death and consequently more significant TRAE. With respect to these concern IgG4 seems to strike a happy medium compared to other IgG Fc isotypes.
The anti-CD47 mAbs in development have different epitopes, meaning they bind to different parts of CD47. The differences in epitopes has resulted in a subset of anti-CD47 mAbs that do not tightly bind to the form of CD47 on the surface of red blood cells (RBC). This type of anti-CD47 mAbis colloquially referred to as RBC sparing. This is an important differentiating factor since cytopenia, a reduction in number of mature blood cells, is the most common severe TRAE for the modality and has slowed the development of anti-CD47 mAbs.
Advantage: Lower potential for ADA
Disadvantage: Potential RBC binding, potential antigen sink, larger MW
Examples: Magrolimab, Lemzoparlimab, AO-176
2) Anti-SIRPα mABs:
While the majority of therapeutics that are being developed for blockade of CD47-SIRPα checkpoint target CD47, a few anti-SIRPα mAbs are being developed that act as SIRPα antagonists. The main advantage of targeting SIRPα, to achieve blockade of the CD47-SIRPα checkpoint, is the limited tissue distribution of SIRPα. This narrow expression profile reduces the potential antigen sink issue related to CD47 antagonists and likely decreases the dose required to achieve maximum receptor occupancy. Additionally, since SIRPα is not expressed on blood cells, treatment with an anti-SIRPα mAb avoids many of the TRAEs that occur when using an anti-CD47 mAb.
However, since SIRPα is primarily expressed in neurons and myeloid cells, triggering Fc-dependent effector function could have a significantly negative effect. So anti-SIRPα mAbs need to have an inactivated Fc region. Inactivation of the Fc region limits the possibility of using anti-SIRPα mAbs as monotherapy.
A major challenge impeding the development of anti-SIRPα mAbs is developing an agent with high enough selectivity. Due to the prevalence of multiple SIRPα alleles, it is challenging to develop an agent that is able to bind the relevant alleles with great enough affinity to blockage CD47-SIRPα. This issue is further complicated by the existence of multiple SIRP homologues. For example, SIRPγ is expressed on the surface of T cells and while its biochemical function is unknown, it may be involved in T cell responses. Therefore, developing an anti-SIRPα mAb that has high affinity for all relevant SIRPα variants and minimal affinity for other SIRP homologs is challenging.

Advantage: Lower potential for ADA, Decreased antigen sink
Disadvantage: Doesn’t reinforce targeting of tumor cells, SIRPα polymorphisms modulate SIRPα-CD47 binding affinity, unknown safety
Examples: ALTA-002, OSE-172
3) SIRPα-Fc fusion:
In general, SIRPα-Fc fusions are composed of the extracellular domain of SIRPα fused to an IgG Fc region. Since CD47 is the native binding partner of SIRPα, SIRPα-Fc fusions are able to selectively bind to CD47. However, the native protein binds to CD47 with affinity (Kd~.5 um) much less than that of a typical mAB. To overcome this limitation, Garcia and coworkers designed variants of SIRPα that bind CD47 with disassociation constants in the picomolar regime.
Fusing the binder to an Fc region has several advantages over use the just the protein fragment. Fist, the presence of the Fc domain slows the clearance of the therapeutic. Second, the Fc domain allows the drug to interact with FcRs and trigger Fc mediated effector functions. Last, the presence of the Fc region can simplify production. Since the Fc domain folds independently, it can help improve the stability of the fusion protein and IgG-Fc affinity resins can be used to purify the product.
Advantage: Smaller MW
Disadvantage: Potential RBC binding, potential antigen sink, greater potential for ADA
Examples: TTI-621, TTI-622, ALX148
4) Bispecific anti-CD47:
Bispecific anti-CD47 includes bispecific antibodies and bispecific fusion proteins. A strong argument for an appropriate use case for this modality has not been made. The main argument is essentially, since anti-CD47 mAbs are being developed for combination therapies, why not start with a bispecific? Additionally, it is has been argued, with little evidence, that targeting anti-CD47 with a bispecific drug may improve safety and efficacy by improving the binding specificity. The advantage of this modality over an antibody cocktail is unclear.
Advantage: Decreased antigen sink
Disadvantage: Greater potential for ADA, very large MW, manufacturing will be more challenging
Examples: SL-172154, TG-1801, IBI322
Which modality to choose?
The space has many different player developing their own take on a CD47-SIRPα checkpoint inhibitors. The modalities that seem the most sensible are: anti-SIRPα mAbs with an inert Fc region, anti-CD47 mAbs that are RBC sparing or have an inert Fc region, and SIRPα-Fc fusions that are RBC sparing or have an inert Fc region. These drugs make the most sense since they seek to limit the potential for serious TRAE.
At this point in the development of CD47-SIRPα checkpoint inhibitors, there are too many unknowns to decide if inert Fc or RBC sparing with active Fc is superior when designing a CD47 antagonist. Inert Fc implies that the drug will only be used in combination with other biologics, since inert Fc usually results in a drug that does not have monotherapy activity, and maximal anti-tumor activity following CD47 blockade requires Fc mediated effector functions. RBC sparing with an active Fc is more likely to have some monotherapy activity and may not require another biologic to maximize efficacy, but there is potential for the “scorpion effect” and competition for Fc receptors when combined with another biologic with an active Fc region.
When designing an anti-SIRPα mAbs it is crucial to develop to take into SIRPα polymorphisms. There are several variants of SIRPα and it is important to design a drug that binds the major variants with high affinity. Additionally, since SIRPα is found on macrophages and dendritic cells, an active Fc region could have a drastically negative effect.
Miscellaneous
Scorpion Effect: In the context of CD47 the scorpion effect refers to when a CD47 antagonist binds to the CD47 on macrophage and the Fc region of that CD47 antagonist interacts with the Fc receptor on the same macrophage.
Macrophage Polarization: Preclinical research done by several indecent workers suggests that inhibition of the CD47- SIRPα checkpoint, either with a CD47 antagonist or SIRPα antagonist, leads to macrophage polarization and increases the ratio of M1 to M2 macrophages. M1 macrophages are commonly characterized as pro-inflammatory and anti-tumoral. M2 macrophages are commonly characterized as anti-inflammatory and pro-tumoral.
Dendritic Cells and the Adaptive Immune System: Dendritic Cells are a type of antigen presenting cell that link the innate and adaptive immune system. Work done by Fu and coworkers suggests that blocking CD47-SIRPα signaling increases tumor mitochondrial DNA in the cytosol of dendritic cells following phagocytosis of the tumor cell. The increase in tumor mitochondrial DNA in the dendritic cell leads to its activation and cross presentation of the tumor antigen to CD8+ T cells, which proliferate and kill the tumor cells.
4. The Parallax View
CD47-SIRPα blockade is a promising strategy to engage the innate immune system and potentially prime the adaptive immune system to treat cancers where CD47 is overexpressed. Some of the therapeutics currently in the clinic could be the innate backbone of cancer immunotherapy.
The field may seem crowded from the outside looking in, but the technologies that are being developed are sufficiently different from each other, with room for multiple winners across different cancers.
While Gilead’s magrolimab has a significant lead on its competitors and is most likely to be the first CD47 antagonist on the market, it has clear limitations. ALX Oncology, Trillium, and I-Mab all have exciting assets in development that will challenge Gilead in the future, which we cover next.
The early clinical data for most CD47-SIRPα checkpoint inhibitors as a monotherapy has been underwhelming. Going forward, these agents will likely be explored for use in combination therapies. This shift toward combination therapy will clarify and provide insight into which strategy is the most promising and which of the potential concerns (e.g. Fc receptor competition and Scorpion Effect) matter most.
Our eyes are on SIRPα-Fc fusion proteins, namely ALX and Trillium, where both companies toot their horn of having a 75kDa molecular weight, which is incredibly important in penetrating solids and exiting the blood into tissues. More importantly, a series of combo trials will unfold going into 2022 and beyond, which will determine if CD47 can be a truly viable innate backbone.
Additionally, CD47 may not be limited to cancer. CD47 likely has applications to broad-spectrum infectious diseases immunotherapy, and it may even be a potential biomarker for the early diagnosis of severe COVID-19.
Finally, keep an eye out for CD24. In 2019, Weissman and co identified CD24 as a promising target for cancer immunotherapy - another ‘don’t eat me’ signal. While early phase clinical data for CD47 antagonists for hematological cancers has been encouraging, much of the early phase clinical data for treating most solid tumors so far has been lackluster. CD24-Siglec-10 is an emerging innate immune checkpoint that is promising for the treatment of solid tumors, such as ovarian & breast. CD24 is overexpressed in many types of solid tumors and has a different expression profile than CD47, where Siglec-10 is expressed by tumor-associated macrophages. Weissman and coworkers demonstrated that blockade of CD24-Siglec-10 checkpoint with mAbs enhanced ADCP in all cancer models that were tested. Expression of CD24 was also found to correlate to response to CD24 blockade and baseline phagocytosis levels, suggesting that CD24 is a potent “don’t eat me” signal. Interestingly, CD24 binding to Siglec-10 results in an inhibitory signaling cascade that is mediated by SHP-1 or SHP-2, in a manner that is similar to CD47 and SIRPα. In short, CD24 might be just as promising as CD47.
5. The Competition
Remember, there will be multiple winners in the CD47 space. Here we cover:
ALX Oncology ($ALXO)
I-Mab Biopharma ($IMAB)
Shattuck Labs ($STTK)
OSE Immunotherapeutics (Euro: $OSE)
Trillium Therapeutics ($TRIL) - a retrospective 🥳
You can find our compiled tracker of clinical trials for TRIL and ALXO here.
Selected Clinical Results for Trillium, ALX, Gilead, I-Mab, and OSE

Sources 1

$ALXO, 2.9B MC - Shooting for Synergy
ALX Oncology is a clinical stage biotech developing therapeutics to block CD47- SIRPα signaling for the treatment of hematological cancer and solid tumors. ALX Oncology is focused on developing therapeutics that will be used in combination with anti-tumor mAbs.
Pipeline:
ALX148 (evorpacept) is a SIRPα-Fc fusion protein with a molecular weight of ~75 kDa. ALX148 consists of the extracellular domain of an engineered variant of human SIRPα fused to an inactivated Fc domain. There are no Fc-mediated effector functions so proposed MOA is blockade of CD47-SIRPα checkpoint to enhance ADCP. It is being developed for use in combination therapies for hematological malignancies and solid tumors.
ALTA-002 is an anti-SIRPα antibody linked a Toll like receptor 9 agonist. ALTA-002 binds the two major human alleles of SIRPα, v1 and v2. The proposed MOA is blockade of CD47-SIRPα checkpoint and activation of TLR9 to generate adjuvant effects and prime the adaptive immune system.

Note: Tallac spun out of ALX/VenBio and is ALX’s private sister company
Science Review:
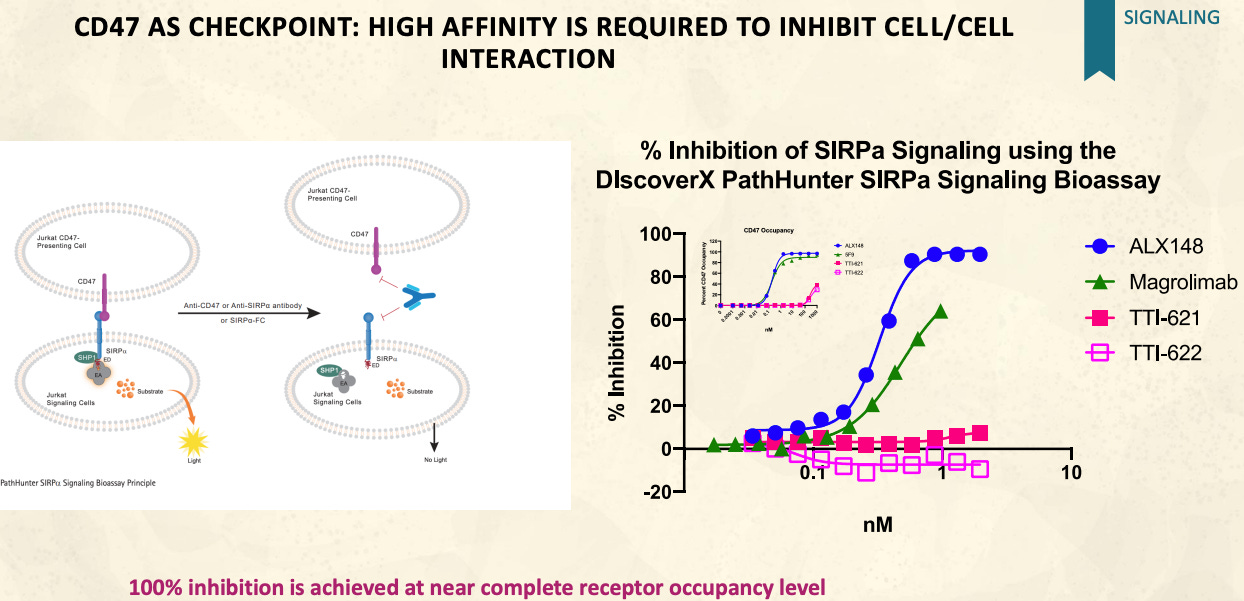
ALX148 is potentially best in class CD47 antagonist that yields very high receptor occupancies with a favorable safety profile. ALX148 is designed to promote ADCP via CD47-SIRPα blockage.
This MOA is being leveraged to pursue a number of combination therapies that may yield synergistic effects. To maximize the ability of ALX148 to be used in combination therapies, ALX has engineered ALX148 to improve the safety profile and to avoid the potential pitfalls, such as the scorpion effect and competition for Fc receptors, that may otherwise arise during combination therapies.
ALX Oncology appears to have a very good handle on the fundamental science and is being transparent. They have developed a number of robust assays and often present all relevant data for their experiments and controls . Based on preclinical work ALX has proposed that by using a strong CD47 antagonist in combination, with therapeutics that have different MOAs, they will be able to exploit synergistic drug effects. Though ALX148 inactive as a monotherapy, data from several early stage clinical trials exploring ALX148 in combination therapies seems to be supporting this hypothesis.
Though TTI-621, TTI-622, and ALX148 are all SIRPα-Fc fusions, ALX148 is a fundamentally different technology from TTI-621 and TTI-622. In our opinion it is not particularly worthwhile to debate the relative merits TTI-621 and TTI-622 versus ALX148. Each of drugs will have its own collection of merits and demerits and potentially different optimal use cases. That being said, ALX148 is clearly superior if the goal is to achieve CD47 blockade.
Clinical Data:
Currently ALX148 is being evaluated in the clinic as a treatment for solid tumors and hematological cancers. ALX148 was first evaluated as a monotherapy for various solid tumors. ALX148 was well tolerated and achieved near complete RO at doses >= 3 mg/kg, but had minimal single agent activity with the 6 of the 24 evaluable patients having a best response of stable disease. Thus, future work was focused on developing ALX148 for combination therapies.
When ALX148 was combined with rituximab to treat patients with relapsed or refractory CD20 positive B cell non-Hodgkin lymphoma it had a response rate that is much greater than rituximab as a salvage therapy. When ALX148 was dosed at 10 mg/kg it had an ORR of 41% with 9 responders, including 4 complete responders, and 6 stable disease out of 22 patients and when ALX148 was dosed at 15 mg/kg it had an ORR of 64% with 7 responders, including 3 complete responders, and 1 stable disease out of 11 patients.
ALX148 at 10 mg/kg was combined with pembrolizumab as a 2nd line or later treatment for head and neck squamous cell carcinoma (HNSCC). It was observed that 4 of 10 checkpoint naïve patients responded to treatment. However, 0 out of 10 patients who had prior checkpoint therapy responded to treatment. For reference, pembrolizumab as a monotherapy for HNSCC has a response rate of 15%. Importantly, the treatment was well tolerated with only 5 TRAE that were grade 3 or greater out of 52 patients.
ALX148 at 10 mg/kg was combined with pembrolizumab as a 2nd line or later treatment for non-small cell lung cancer (NSCLC). The therapy had 1 partial responder out of 20 patients.
ALX148 at 10 mg/kg was combined with pembrolizumab and chemotherapy as a 1st line treatment in checkpoint naïve patients for treatment of head and neck squamous cell carcinoma (HNSCC). This resulted in 3 out of 4 patients responding. The benchmark for pembrolizumab and chemotherapy for this indication and patient group is 36%. Though the sample size is very small for the ALX148 combination, this result is exciting.
ALX148 is being evaluated as a 2nd line or later treatment for HER2 positive gastric cancer. When combined with trastuzumab, an anti-HER2 mAb, it yielded 4 partial responders out of 19 patients. When combined with trastuzumab, paclitaxel, and ramucirumab, an anti-vascular endothelial growth factor receptor 2 mAb, it yielded 13 responders out of 18 patients. For reference trastuzumab in combination with paclitaxel has a response rate of 33% and ramucirumab in combination with paclitaxel has a response rate of 28%.
Overall the clinical data that has been released by ALX is compelling. The rituximab and ALX148 combination therapy for NHL and the trastuzumab, paclitaxel, ramucirumab, and ALX148 combination therapy for gastric cancer both seem very promising. The data for ALX148 used in combination for HNSCC is exciting for checkpoint naïve patients, but is not encouraging for patients with prior immune checkpoint therapies.
What we like:
Inactive Fc receptor has many potential benefits. By inactivating the Fc receptor it eliminates potential issues with combo therapies using an anti-tumor mAb for competition of Fc receptors. It improves the safety since it will not kill RBC due to Fc-mediated effector functions while retaining the benefits of the Fc structure. This is a huge plus.

Uses SIRPα variant that binds CD47 with very high affinity, similar to that of a mAb. This approach means that ALX148 will have a high receptor occupancy and be able to block the CD47-SIRPα checkpoint.
Focusing on combination therapy makes sense since the ORRs for existing immune checkpoints and existing CD47 antagonists as monotherapies are low, so in the long run they will likely end up being used in combination therapies.
In their clinical trials, the control arm or reference combination is the standard of care. This makes the effect of their combo therapy we clear and allows them to directly identify synergies. Also, it means that ALX is not playing the get a high ORR game with a combo that lacks an appropriate control.
They do good science. The reasoning behind the design and development of ALX148 and ALTA-002 makes sense. Their preclinical and clinical data is presented in a very clear manner and they present the relevant controls.
Development of ALTA-002 is a good move. SIRPα is more narrowly distributed so removes potential antigen sink issue. SIRPα is over expressed in a several cancers including head and neck cancer. The choice of conjugate is very sensible and takes advantage of SIRPα’s expression profile.
What we dislike:
ALX is not upfront that they have to pursue combo therapy with their technology because there is no path forward for any iteration of their technology as monotherapy. ALX377 (essentially ALX148 fused to a wild type IgG1 Fc domain) likely isn’t safe and ALX148 isn’t effective as a monotherapy. In a murine model treatment with ALX377 resulted in 34% decrease in RBC count, 70% decrease in platelet count, and 67% decrease in white blood cell count three days post-dosing compared to baseline counts. This demonstrates that their drug is not RBC sparing.
They spend too much time slinging mud, especially at Trillium. We assume part of this is also to make a case about how their technology is sufficiently different from prior art described in patent US 10,907,209.
At the end of the day, maximum antitumor efficacy from blockade of CD47 requires Fc-Fc receptor interactions. Even though ALX148 binds super tightly to CD47, it is going to need to be combined with a biologic with an active Fc. This limits the types of combinations that ALX can pursue.
Strategy & Management Review:
AML is the only overlapping indication between TRIL and ALXO. We believe this was perhaps deliberate choice made by Trillium to not (1) squarely focus on solids and (2) fight directly against ALXO. This in turns gives ALXO a boost as it retains its daring title as the player most daringly tackling solids head-on (HNSCC, Gastro/Gastric, Breast)
Hidden from the limelight is Board Chairman and Co-Founder Corey Goodman who is a bit of genius. He was Chair of both Renovis & Labrys, both of which were acquired. Also the co-founder of seven other biotechs. Read more here. He is a sort of shadow-CEO with lots of say given that Jaume Pons & ALXO was incubated out of VenBio.
At the expense of not having an active ‘eat me’ signal due to its inactive Fc, it has more combo opportunities with different drugs & companies, as testified by its ongoing trials with Merck, Lilly, and Zymeworks
HNSCC, HER2+ Breast, Gastro/Gastric are solids, and consequently bigger indications by new populations p.a. and overall market size. It is also less crowded vs hemes. Current SOC are deeply unideal and ALXO has a real chance to run successful triplets with chemo + ICIs
They are relatively more ahead in their Phase 2 trials versus competitors, which have higher N-counts and also feature a global gastric trial
General excitement around their data and future company prospects and its positioning as a solid-combo partner that can create synergy
% Institutional Ownership (ranked by %): Venbio (24%), LSV (11.7%), Vivo Capital (10%), Logos (7.8%) – likely sold all given Board Member resigned from $ALXO, Wellington (7.2%), Vanguard (3.9%), Fidelity (3.1%), BlackRock (3.1%), Janus (3%), State Street (2.6%), Redmile (2.2%), Cormorant (2.2%)
Significant Q2 Adds: Eventide (350K shares) and Wellington (235K shares)
Insider Activity: Nearly 80K shares were sold in August – or about 0.5% of the float. 8/17: CFO sold 10K shares in the 60s (~$650k). 8/19: CEO sold 9K shares in the 60s (~$500k). 8/23: CFO sold 60K shares in the 70s ($4m+)
Financial health:
40.3m shares outstanding with a 17.35m float (43%)
~14.3% held by insiders, ~83% held by institutions
Cash and cash equivalents: an ample $410m with a cash burn of $15m/quarter, giving an ample 6.5 year runway
Estimated enterprise value: ~$2.4B
The case for an ALXO buyout:
ALXO is the most attractive candidate leftover for a CD47 buyout. The main reason is two-pronged: (1) it is tackling solids head-on and the heme market is led by FTSV and TRIL, and (2) it is the most robust SIRPα-FC fusion protein left on a market saturated by somewhat questionable mAbs.
We know that GILD made four separate offers to FTSV, from $57.5 to $64.5 to $66 to $77 to $95.5, where FTSV at one point demanded a $68.5 minimum offer (valuing it then at $3.3B). Whats interesting was that we know there was another bid for $FTSV that was at least $77 on Feb 28 (valuing it at $3.7B), which forced GILD to place a higher counteroffer. Read it all here. These may or may not overlap with Parties A, B, C in $TRIL’s 14A.A few takeaways from this is that $ALXO is at $3.0B and is superior to GILD in terms of solids. So there is at least a 20% valuation gap to FTSV’s other known bidder’s revealed WTP of $77.
Merck seems to be the leading BP name floated around as a suitor for ALXO. Merck’s recent investor presentation states “Ex-U.S., 27% growth driven by continued uptake in lung and ongoing launches in HNSCC.” Given that the HNSCC market is absolutely huge in Asia, we see this as a nice fit between ALXO and Merck especially since some of ALXO’s trials will be global (gastric/gastro). Merck has also been partnered with ALXO since September 2020, whereas Lilly only inked its first partnership in June this year. While Merck expressed interest in M&A for companies with monotherapy results that are pre-commercial, we believe that the partnership gives Merck a strategic initial foothold into the company.
Another argument that Merck needs CD47 is that future Keytruda pricing could be more influenced by the crowded PD-(L)1 space and fast-followers from China. Owning ALXO would at least secure at differentiated CD47 player that is not a CD47 monoclonal antibody but a fusion protein that works well in tandem with Keytruda. In other words, ALXO also holds scarcity value as a well-positioned combo partner for big pharma.

$IMAB, ~$5.2B MC - The China-grown mAb leader
I-Mab is a Chinese clinical stage biopharma developing biologics for immuno-oncology and autoimmune diseases. Their pipeline includes Lemzoparlimab (also known as TJC4 or TJ011133, or lemzo for short), a human anti-CD47 mAb of the IgG4 isotype that is red blood cell sparing. It is proposed that the RBC sparing property is due a RBC specific glycosylation of CD47 that prevents Lemzoparlimab binding (glycosylation of the epitope). The MOA is presumedly blocking CD47-SIRPα signaling to promote ADCP and using CD47 to target and subsequently eliminate cancer cells via Fc-mediated effector functions.
I-Mab is focused on developing Lemzoparlimab for solid tumors and hematologic cancer. Data from I-Mab’s Phase 1 trial evaluating Lemzoparlimab as a monotherapy for relapsed or refractory solid tumors and lymphoma has been published. Dosing was escalated to 30 mg/kg and was well tolerated with only a single TRAE of grade 3 or higher and dose limiting toxicities were not observed. However, anti-drug antibodies were observed in 5 out of 20 patients. A weekly dose of 20 mg/kg or higher resulted in saturating receptor occupancy. Out of 20 patients there was 1 partial responder. Somewhat excitingly, the partial responder was one of the three subjects dosed at 30 mg/kg. The patient had metastatic melanoma and had failed prior treatments with checkpoint inhibitors, nivolumab and ipilimumab.
I-Mab and AbbVie partnered on Lemzoparlimab in September of 2020. AbbVie agreed to give I-Mab $180 million USD upfront, $20 million in milestone payments based on the Phase 1 results, and up to $1.74 billion in success based milestone payments. Additionally, I-Mab retains the rights to Lemzoparlimab in China and receives low to mid teen percentages of global net sales outside of China.

What we like:
AbbVie is primarily responsible for manufacturing the global supply of Lemzoparlimab.
RBC sparing so avoids potential antigen sink and increases tolerability
High receptor occupancy was observed at a dosing that is well tolerated.
What we dislike:
Anti-Drug Abs were found in 5 out of 20 patients
Lemzoparlimab had a low response rate as a monotherapy (1 out of 20)
In terms of outlook, I-Mab will likely derive most of their CD47 value from China. Currently, there are three major trials:
In combination with rituximab for NHL, where they submitted an abstract for ASH as they were excited about the ongoing trials. They are also now expanding and doing a China-side trial in September. They think the current clinical trial can lead to a registrational study for NHL in 2022.
In combination with AZA for AML/MDS. This P2 trial is based in China where all patient enrollment is expected to be done by YE and to segue into a registrational trial in 2022.
In combination with pembro for solid tumors. They anticipate Q4 21 or Q1 22 as the earliest time to report efficacy. China has received approval for a Phase 2 trial targeting more than 1 solid tumor.
In total, I-Mab has 86 patients dosed in both US and China where no priming dose is needed throughout the trials, with 1~2 pivotal studies anticipated to be released in 2022. In the Aug 31 earnings call, management repeatedly iterated the safety profile of lemzoparlimab and cited priming doses were not needed because of safety. They are prepared to release data in November, if not earlier.
We note that they “observed very encouraging clinical efficacy signals” that are “among the best efficacy signals seen so far with clinical stage CD47 antibodies around world” in their AML/MDS combo trials with AZA.
Strategy, I-Mab likely isn’t a buyout candidate given its Chinese background and ambitions to scale up into a fully vertically-integrated firm, but management did note that its commercial strategy is to focus on hematological malignancies and to have lemzoparlimab become a backbone drug for AML/MDS. This would be especially important as they want to become the first CD47 antibody commercialized in China and to fuel AbbVie’s global success with it. We note that AbbVie’s manufacturing capabilities are among the best in BP. We are also big fans of I-Mab’s management and believe I-Mab is undervalued given its many other promising clinical assets.

$STTK, ~$900m MC - Too Big to Handle?
Shattuck Labs is a clinical stage biotech focused primarily on developing biologics for immuno-oncology using their agonist redirected checkpoint (ARC) platform. The ARC platform is based on linking the extracellular domains of an immune checkpoint receptor and a tumor necrosis factor (TNF) superfamily ligand to an inert Fc. The ARC compounds self-assemble to become a hexamer of checkpoint receptors and two trimers of TNF ligands.
SL-172154 is the extracellular domains of SIRPα and CD40 ligand fused to an inert IgG4 Fc. SL-1721534 binds immobilized CD47 and CD40 with affinities of 630 pM and 4.7 nM, respectively. The vision of SL-1721534 is a therapeutic that bridges adaptive and innate immunity. It is being developed for solid tumors and hematological malignancies as monotherapy and is currently in a Phase 1 trial for Ovarian Cancer and a Phase 1 trial for cutaneous squamous cell carcinoma (CSCC) and head and neck squamous cell carcinomas (HNSCC).
Preclinical data using non-human primates (NHP) demonstrates that SL-172154 is likely going to be well tolerated since no evidence of hemolysis, thrombocytopenia, or liver enzyme elevation was observed. During dose-escalation studies with NHP a CD47 receptor occupancy of ~80% was observed 1 day after infusion at 10 mg/kg or greater was ~60% 7 days after infusion. Phagocytosis activity assays were completed, using several tumor cell lines, that suggest that SL-172154 increases phagocytosis.
Given that wildtype SIRP does not bind CD47 tightly, we would not expect that SL-172154 work well as a CD47-antagonist. However, Shattuck reported a dissociation constant that is much lower than expected, given that they are using the extracellular domain of wild type SIRPα. Thus, it is not clear why SL-172154 would bind CD47 ~1,000 times tighter than TTI-621 or TTI-622. While the RO for SL-172154 in NHP seems decent, ALX’s data and Trillium’s data taken together show that 60-80% RO is probably not enough and >90% RO is needed to block CD47-SIRPα signaling. We are not convinced that SL-172154 will be effectively blockade the CD47-SIRPα checkpoint.

What we like:
Structurally distinct from existing biologics.
Platform does not require complex screening since it mixes and matches known ligands.
TNF receptors usually have ligands that readily form trimers. Since the ARC molecules self assembles to form a trimer of the TNF ligand this modality may activate the TNF receptor better than previous technologies.
SL-172154 probably will probably be well tolerated in clinical trials.
What we dislike:
We are unconvinced that SL-172154 will have monotherapy activity or effectively blockade the CD47-SIRPα checkpoint. Since the Fc domain is inert, if it does not block CD47-SIRPα signaling, SL-172154 is going to be useless.
Their phagocytosis activity assay data is not super convincing and we would have liked to see concentration dependence with respect to the biologic.
SL-172154 has a molecular weight in excess of 450 kDa so it will likely have limited tissue penetrating ability. It is six times mass of ALXO and Trillium’s assets.
EUR: $OSE, 230M MC - Allele restricted SIRPα-mAb?
OSE Immunotherapeutics is a French clinical stage biotech developing therapeutics for immuno-oncology and autoimmune diseases. They have a diversified pipeline that includes OSE-172 (BI 765063), an anti-SIRPα mAb that is being developed in collaboration with Boehringer Ingelheim since 2018. The deal structure with Boehringer Ingelheim includes 35 million euros that have already been received, up to 1.1 billion euros in future milestone payments, and high single digit to low teens royalties on global sales.
OSE-172 is a humanized IgG4 mAb that binds SIRPα V1 with high affinity and binds SIRPα V2 with low affinity. Additionally there is no cross reactivity with SIRPγ, which is expressed on the surface of T cells and may play a role in antigen proliferation.
Based on OSE’s publications it seems likely that OSE-172 will only be able to blockade CD47-SIRPα in patients who are SIRPα V1/V1 homozygous. Unfortunately, this is a significant flaw of OSE-172 and dramatically limits the usefulness of OSE-172.
OSE-172 is in clinical trials as a monotherapy for advanced solid tumors. Phase I data has been published a revealed that it was well tolerated and resulted in sustained receptor occupancy saturation. There were a total of 50 patients (26 V1/V1, 24 V1/V2) with various advanced solid tumors. One patient with hepatocellular carcinoma (HCC) with liver and lung metastases and 7 prior lines of therapy showed a durable partial response maintained for 27 weeks treatment. A phase 1 clinical trial evaluating OSE-172 in combination with BI 754091, an anti-PD-1 mAb, for solid tumors is ongoing.
Keep an eye out for their ESMO 2021 update in mid-September.
What We Like:
OSE-172 does not bind SIRPγ.
Focused on solid tumors.
What We Don’t Like:
OSE-172 does not appear to bind the V2 variant of SIRPα tightly, so it cannot be used as a treatment for most patients.
It only had a single partial responder as monotherapy. ORR was only ~4% for SIRPα V1/V1 homozygous.

$2.26B Buyout ($18.5/share) – Although acquired, we still wanted to provide a review & retrospective on what was our top pick, where our CD47 portfolio was 70% Trillium. We have been following $TRIL since 2017.
Pipeline:
1) TTI-621 is a SIRPα-Fc fusion protein with molecular weight of ~75 kDa. TTI-621 is the extracellular domain of human SIRPα fused to IgG1 Fc domain. The IgG1 isotype demonstrates a high affinity for Fc receptors and is a potent activator of Fc-mediated effector functions (ADCC, ADCP, and CDC). MOA is proposed to include the recruitment of macrophages and natural killer cells to eliminate the cancer cells. TTI-621 is being developed as a monotherapy and for use in combination therapies.
2) TTI-622 is a SIRPα-Fc fusion protein with molecular weight of ~75 kDa. TTI-622 is the extracellular domain of human SIRPα fused to IgG4 Fc domain. The IgG4 isotype demonstrates a weak affinity for most Fc receptors and is a weak activator of Fc-mediated effector functions. MOA is proposed to include the recruitment of macrophages to eliminate the cancer cells. TTI-622 is being developed for use in combination therapies.

Clinical Data:
A phase I trial have been conducted to evaluate TTI-621 as monotherapy and in combination with either rituximab (anti-CD20) or nivolumab(anti-PD-1) for relapsed or refectory hematologic malignancies. For the indication of diffuse large B-cell lymphoma (DLBCL) the ORR was 29% (2/7) for TTI-621 as a monotherapy and 21% (5/24) for TTI-621 plus rituximab. Due to the relative small sample size it is difficult to say anything conclusive, but it does not seem like the combination improved the ORR. It is important to note that while magrolimab and ALX148 lack efficacy as a monotherapy, each had synergistic effects when combined with rituximab for treatment of relapsed or refractory CD20 positive B-cell non-Hodgkin’s lymphoma yielding an ORR of 50% and an ORR of 70%, respectively. For reference, rituximab monotherapy as a salvage therapy has an ORR of 10-15%.
Patients with Hodgkin’s lymphoma received either TTI-621 as a monotherapy TTI-621 and nivolumab as a combination therapy. Out of the 20 patients that received the monotherapy there was only 1 partial responder. Out of the 4 patients that received the combination there was 1 complete responder and 1 partial responder. Though nothing definitive can be said, it seems like combining TTI-621 with an immune checkpoint inhibitor has a synergistic effect.
Attributing the lack of synergy observed for the TTI-621 and rituximab combination to the scorpion effect or competition over Fc receptors is not very convincing since magrolimab has an active Fc. These results could instead suggest that the MOAs of TTI-621 and rituximab are sufficiently similar in this context, such that combining them does not improve efficacy. Along this line, Trillium’s ADCC assays comparing TTI-621 and rituximab using human NK cells and B-cell lymphoma targets had similar activities.
Maximum tolerated dose (MTD) assigned to TTI-621 in this trial was 0.2 mg/kg. At dose of 0.2 mg/kg the median CD47 RO is 34%. Though it is likely that 0.2 mg/kg is an underestimate for the MTD, the median RO at 0.5 mg/kg is still only 66%. While some patients had ROs close to 90%, it seems like the vast majority of patients had ROs that were too low to block CD47-SIRPα. This argument seems reasonable since TTI-621 is a relatively weak binder of CD47 and most CD47 antagonist see saturating ROs at several mg/kg.
The above referenced clinical data may support the hypothesis that at the dose of TTI-621 used in the trial, the benefit that is observed is the result of targeting CD47 as a tumor antigen and not from CD47-SIRPα blockade. If this is true, it is not a negative. Instead, it could be a feature that differentiates the technology and allows Trillium to pursue alternative combination therapies.
Additionally, a clinical trial evaluating TTI-622 as monotherapy is ongoing and interim data shows that TTI-622 is well tolerated. Doses of up 18 mg/kg have been administered and Phase 1b/2 studies will start at 8mg/kg. Since the maximum dose of TTI-622 is much greater than the maximum dose of TTI-621, it is possible that TTI-622 is able to achieve a great enough RO to function as a CD47-SIRPα blockade. RO data demonstrates a dose-dependent increase in RO of peripheral blood T-cells and at 18 mg/kg mean RO is near 80%, supporting this possibility.

What we like:
TTI-621 and TTI-622 do not bind CD47 on red blood cells with a high affinity. Anemia and thrombocytopenia are common treatment related adverse events observed following treatment with CD47 mAbs. So sparing RBCs could improve the safety profile. Though, thrombocytopenia was observed in clinical trials with TTI-621 in appears to be transient.
TTI-621 and TTI-622 have monotherapy activities and complete responders have been observed. By demonstrating monotherapy efficacy instead of trying to identify synergies Trillium was able to meaningfully differentiate their technology from ALX’s technology.
TTI-621 and TTI-622 have an active Fc domain. Therefore, they can still leverage Fc-mediated effectors functions in combination with small molecule drugs.
Smaller molecular weight, compared to existing anti-CD47 mAbs, may increase the tissue penetrating ability of TTI-621 and TTI-622.
What we don’t like:
CD47 receptor occupancy is not very high. While wildtype SIRPα binds CD47 selectively (Kd ~ .5um), it binds CD47 much weaker than a mAb. Intuitively, this makes sense since SIRPα-CD47 binding in vivo is transient. Since TTI-621 and TTI-622 use wildtype SIRPα domains it is possible that they are not able to sufficiently block CD47-SIRPα signaling and efficacy that is observed is a result of Fc-mediated effectors and using CD47 as a tumor antigen.
During Trillium’s research day they published phagocytosis assay data of an AML cell line comparing TTI-621, TTI-622 and SIRPα-Fc fusion with an inert IgG4. The assay revealed that the SIRPα-Fc fusion with an inert IgG4 does not result in ADCP, which is to be expected. They also presented data from two murine models demonstrating potential synergies between TTI-621/TTI-622 and Daratumumab or Cetuximab. Though this is data is exciting, it would have been clarifying if Trillium used the SIRPα-Fc fusion with an inert IgG4 from the phagocytosis assays as an additional control, since this data would demonstrate whether TTI-621 and TTI-622 are able to enhance ADCP by CD47-SIRPα blockage.

Potential competition for Fc receptor between TTI-621/TTI-622 and an anti-tumor antibody used in combination could limit TTI-621/TTI-622 applicability as combo agent. This concern is potentially supported by results from Trillium’s Phase I study of TTI-621 in combination with rituximab in patients with relapsed or refractory hematologic malignancies
Potential for the “scorpion effect.” While overexpressed on cancer cells, CD47 is still found on the surface of macrophages. TTI-621/TTI-622 may bind to CD47 on the macrophages and recruit the Fc receptor on the same macrophage.
Known unknowns:
How much receptor occupancy is required to block CD47? Lower RO makes sense given the affinity of SIRPα for CD47. In trials for TTI-621 and TTI-622 as a monotherapy for certain lymphomas there is evidence of a dose dependent increase in receptor occupancy that results in RO of ~>70%. Is that RO sufficient to block CD47-SIRPPα and promote ADCP? We think probably not.
Should competition for Fc receptors be a real concern? There is limited meaningful direct evidence supporting competition of the Fc receptors. Preclinical work done by Trillium suggests that this will not be an issue but, work done by ALX Oncology using a version of TTI-621 and TTI-622 they attempted to produce, based on the literature, suggests it may be an issue.
Could TRIL have had a better offer? Overall biotech macros were depressed, and likely there was no competing bids, especially given the first $25m infusion (breakthrough investment) from Pfizer last year. Lastly, we note that Trillium’s heavy emphasis in dosing hemes first (LMS was 1st dosed only end of June – with Ovarian and Solid #3 still TBD) meant that solid results held substantial uncertainty. Pfizer’s acquisition call also suggests that they were most excited to position Trillium more strategically in their heme products rather than wedging it into its solids portfolio (still possible). Pfizer definitely got a good deal given that Trillium was at a near 52-week low, especially as it has huge amounts of cash and the deal was less than 5% of Pfizer’s massive $45B annual revenue. Furthermore, the Schedule 14A filing suggests a new Party A was a smaller pharma that was ultimately unable to engage in a strategic partnership, and that Party B & Party C ultimately demonstrated no interest in buying Trillium. In short - no bidding war unlike Forty-Seven.
We wrote the below on $TRIL pre-acquisition:
Carving out its turf. TRIL has picked primarily heme indications and LMS, ovarian, and one other TBD as the first solids, for good reason. This is because it is looking to establish its own turf by carving out indications that it believes it can vastly improve on the SOC. While some of these indications may not have the highest new patients / yr or be the largest market size, Trillium has picked the right places to play that will allow it to most optimally (1) capture market share in those indications and (2) expand most quickly into adjacent and periphery indications. We are confident that TRIL’s management has balanced risk (by not squarely focusing on solids), payoff (proven success in heme), and clinical potential (with a new CMO and SAB).
No time to lose. TRIL is plucking low hanging fruit to make up for lost time the past few years – if it can show even good progress in ovarian or leiomyosarcoma- then it can move laterally to similar subtype indications or move up lines of therapy with more ease. It’s setting itself up for clinical success and this has often been overlooked. While there seemed to be disappointment that TRIL wasn’t jumping into solids right away, it should be reminded that $TRIL went through deep growing pains the past few years to find the RP2D, especially the past year of little to no clinical progress given the excitement about monotherapy being the cure-all solution, which was never the intention either.
Don’t overlook monotherapy. While this will be discussed in the science section, it should be noted that monotherapy is a huge part of Trillium’s strategy in how it intends to position itself both on the market and with combo drugs. Jan cited this fact again and again during the R&D day, and the fact that virtually all top oncology drugs had monotherapy activity.
People. CEO Jan Skvarka remains a hidden and often underappreciated gem. Since joining only in September 2019, he salvaged a sinking ship and completely turned the company around as an ex-Bain partner specializing in life sciences. Board member Scott Myers also has a reputation for biotech M&A and his track record speaks for itself. Trillium also has a transactional advisor (Jade Brown). Let’s not also forget the previous M&A activity of Dr. Catherine Mackey and Paulo Pucci. (i.e. keep an eye out for all these folks).
Selected Links and further readings:
I-Mab Aug 31 Call transcript: https://www.nasdaq.com/articles/i-mab-imab-q2-2021-earnings-call-transcript-2021-08-31
(NSFW!) We found Swiss Biopharma Med’s CD47/TNFa mAb, sB24M, quite fascinating as illustrated in this deck (NSFW patient photos)
Next month: CAR-NK or synthetic lethality... or let us know what you’d like to read about!
Disclaimer: Biotech investing is inherently risky. Our post is for informational purposes only, published to the best of our knowledge and understanding. We accept no liability for any potential direct or indirect losses as a result of our research and views. The reader bears full responsibility for their investment decisions. We reserve every right to adjust our positions without notice.
Enjoyed this? Subscribe for company-specific deep dives or give it a share.
https://s22.q4cdn.com/183592819/files/doc_news/2021/08/Project-Argentum-Press-Release_FINAL-FOR-PDF3.pdf
https://alxoncology.com/wp-content/uploads/2020/06/ALX-ASCO-Poster-May20.pdf
https://www.sec.gov/Archives/edgar/data/1810182/000156459020053588/alxo-ex991_6.htm
https://ir.alxoncology.com/static-files/16e6c9e0-e0fa-425d-a5b8-b6fd00431c74
https://ir.alxoncology.com/news-releases/news-release-details/alx-oncology-announces-new-data-aspen-01-phase-1b-study-alx148-0
https://alxoncology.com/wp-content/uploads/2020/11/TMKim-et-al-ASH-3016-Dec2020.pdf
https://www.askgileadmedical.com/downloads/pdfs/conference-materials/oncology/asco-2021/Manero_et_al.pdf
https://www.gilead.com/news-and-press/press-room/press-releases/2020/12/magrolimab-demonstrates-clinical-responses-in-ongoing-phase-1b-trial-of-previously-untreated-acute-myeloid-leukemia-patients
https://www.inoncology.com/sites/default/files/poster/asco21_champiat_1443.1_sirpa_mono_poster.pdf
https://www.i-mabbiopharma.com/userfiles/images/2019-10-30/sitc2020pdf.pdf